(Co-)first-author papers
Pairtree: Fast cancer phylogeny reconstruction using multiple cancer samples
March 2021bioRxiv
Wintersinger JA, Dobson SM, Stein LD, Dick JE, Morris Q.
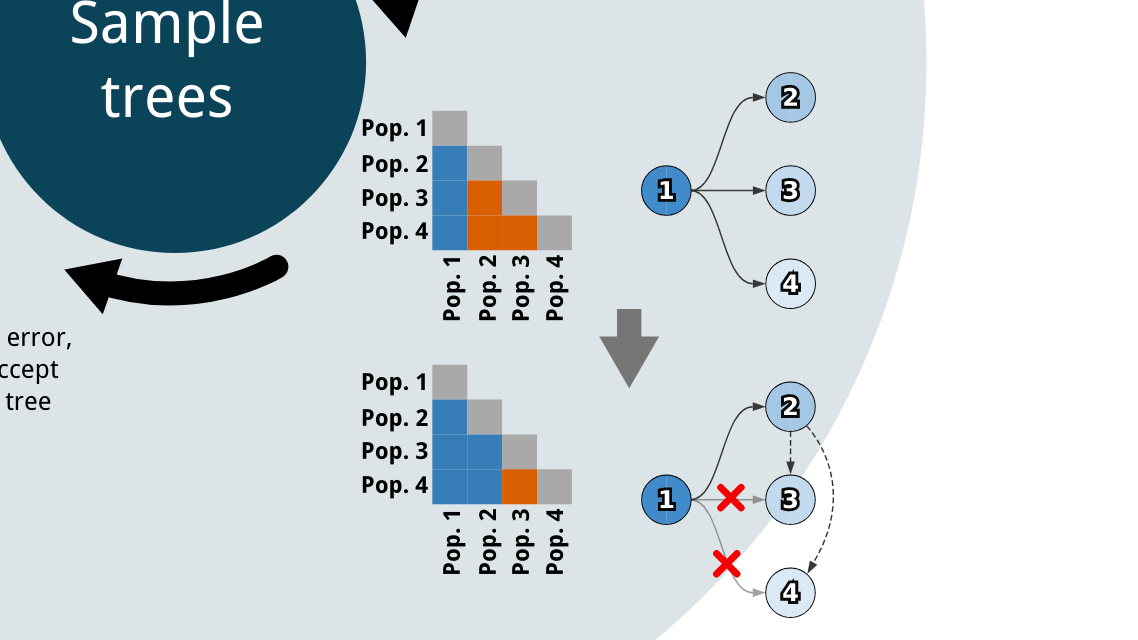
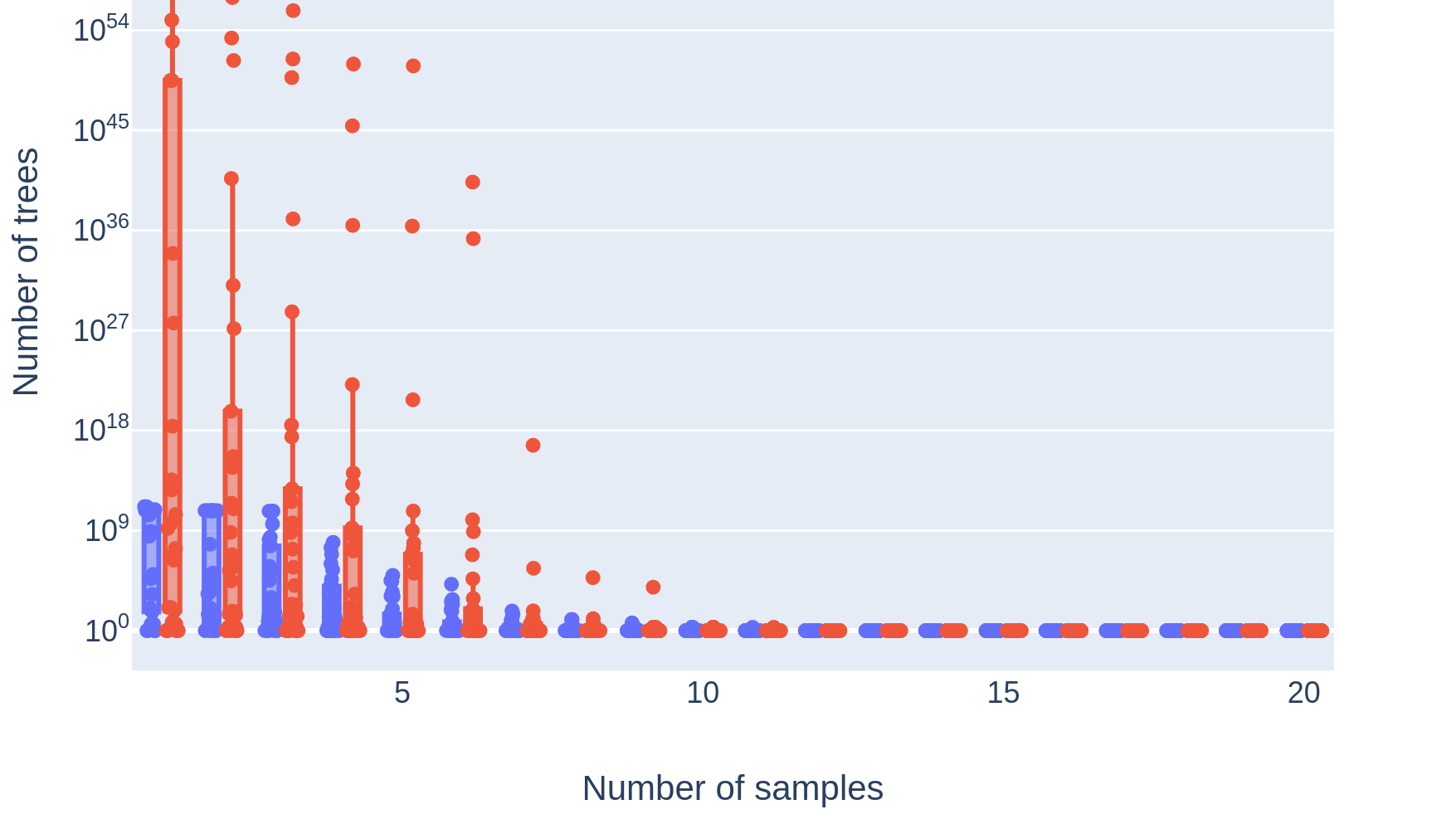
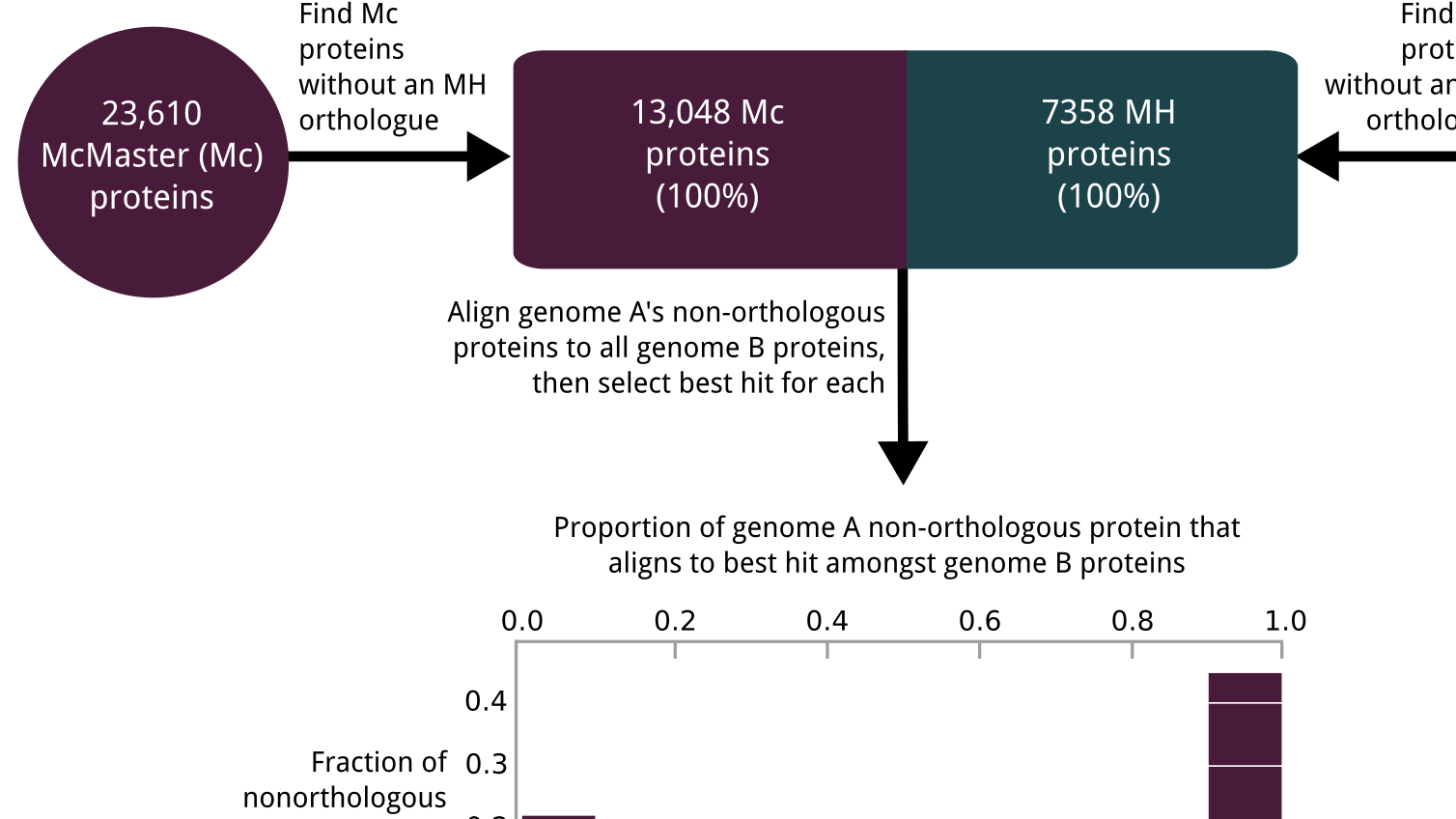
Cancer clone trees depict the genetically heterogeneous cell populations composing an individual patient's cancer, and have important biological and clinical applications. While previous algorithms have been developed for building clone trees from bulk DNA sequencing data, none could scale to settings with more than a few subpopulations or tissue samples. Work we undertook with John Dick's lab, resulting in the Dobson et al. paper described below, necessitated the development of a new approach. Pairtree represents the bulk of my PhD research, and underlies several projects I contributed to in other research groups. Fundamental to this project was the recognition that, whether in real or simulated settings, the observed data often permit multiple equally consistent solutions. Pairtree attempts to capture this inherent ambiguity in the solutions it presents, and to provide tools for understanding the degree of certainty underlying each part of the solutions it produces. Previous methods typically did not acknowledge this ambiguity in their benchmarks on simulated data, nor provided tools for exploring this uncertainty on results generated from real data. The Pairtree implementation reflects an enormous amount of effort over several years to develop a mature, robust method that will be of use to the cancer genomics community. Most of the codebase is in Python, with performance-critical numerical algorithms implemented in Numba, and a web-based interactive visualization suite implemented in JavaScript.
Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes
April 2021Cell
Dentro SC, Leshchiner I, Haase K, Tarabichi M, Wintersinger J, Deshwar AG, Yu K, Rubanova Y, Macintyre G, Demeulemeester J, Vázquez-García I, Kleinheinz K, Livitz DG, Malikic S, Donmez N, Sengupta S, Anur P, Jolly C, Cmero M, Rosebrock D, Schumacher SE, Fan Y, Fittall M, Drews RM, Yao X, Watkins TBK, Lee J, Schlesner M, Zhu H, Adams DJ, McGranahan N, Swanton C, Getz G, Boutros PC, Imielinski M, Beroukhim R, Sahinalp SC, Ji Y, Peifer M, Martincorena I, Markowetz F, Mustonen V, Yuan K, Gerstung M, Spellman PT, Wang W, Morris QD, Wedge DC, Van Loo P, PCAWG Evolution and Heterogeneity Working Group and the PCAWG Consortium.
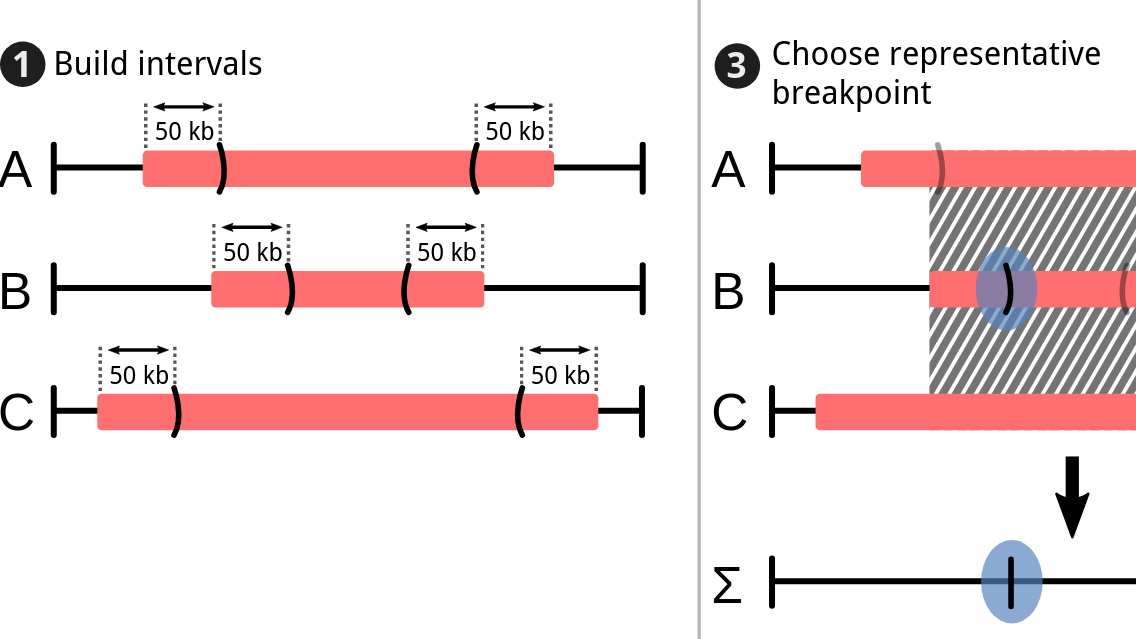
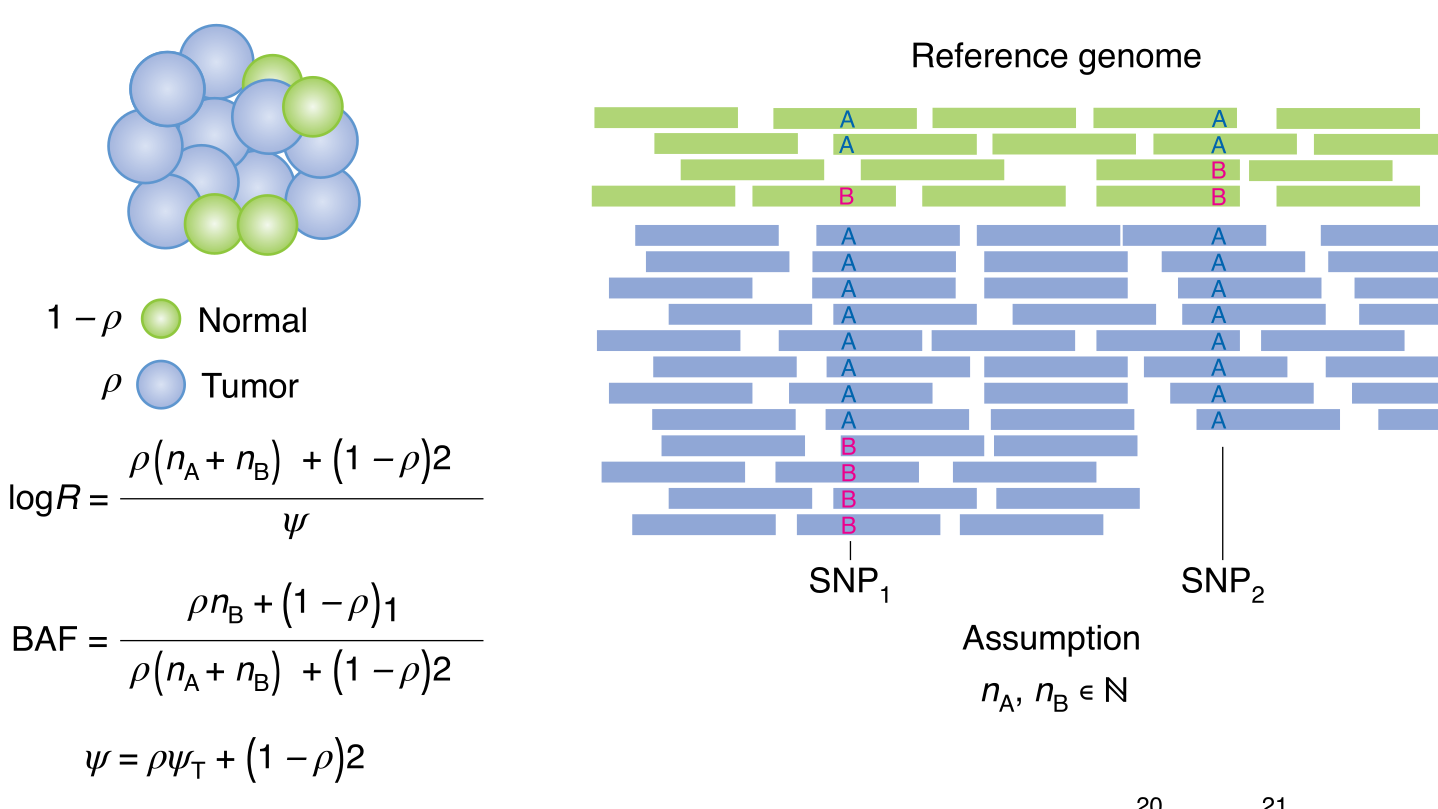
During the early part of my graduate work, I contributed to the Pan-Cancer Analysis of Whole Genomes (PCAWG), an international effort to use whole-genome sequencing to study 2658 newly sequenced cancers, where I was part of the Evolution and Heterogeneity working group. My primary contribution was to build consensus profiles of copy-number aberrations (CNAs) for each of the cancers. The working group had multiple individual methods for characterizing the CNAs for each cancer, but their results differed radically with respect to the genomic breakpoints where copy-number status changed, the total copy number for each genomic segment, the allele-specific copy number, and even whether there was enough information to infer copy-number status at a given locus. I developed a benchmark suite that revealed considerable differences in precision and recall for each method relative to a ground-truth dataset, then devised a tiered consensus scheme that first produced consensus genomic breakpoints where copy-number status changed, and then used the results of individual methods on each resulting segment to infer consensus CNAs, drawing on the strengths and weaknesses of each method revealed by the benchmark. This work was essential for both of the papers our working group produced, as it lay the groundwork for characterizing the evolutionary history of each cancer. The first ten authors on this paper, including me, are co-first authors.
Notable papers
Projects to which I made major contributions
Reconstructing tumor evolutionary histories and clone trees in polynomial-time with SubMARine
January 2021PLOS Computational Biology
Sundermann LK, Wintersinger J, Rätsch G, Stoye J, Morris Q.
SubMARine is an approach for constructing a data structure that concisely summarizes all possible evolutionary histories for a cancer in polynomial time. Dr. Linda Sundermann and I worked on SubMARine through her postdoctoral work. I developed a prototype version of the algorithm, and Dr. Sundermann greatly expanded and refined the concept. For SubMARine, I created the simulated data framework, as well as an algorithm for efficiently enumerating all possible tree structures from noise-free observations of mutation data. I also analyzed experimental results and created figures for the paper.
Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy
January 2021Cell
Rehman SK, Haynes J, Collignon E, Brown KR, Wang Y, Nixon AML, Bruce JP, Wintersinger JA, Mer AS, Lo EBL, Leung C, Lima-Fernandes E, Pedley NM, Soares F, McGibbon S, He HH, Pollet A, Pugh TJ, Haibe-Kains B, Morris Q, Ramalho-Santos M, Goyal S, Moffat J, O’Brien CA.
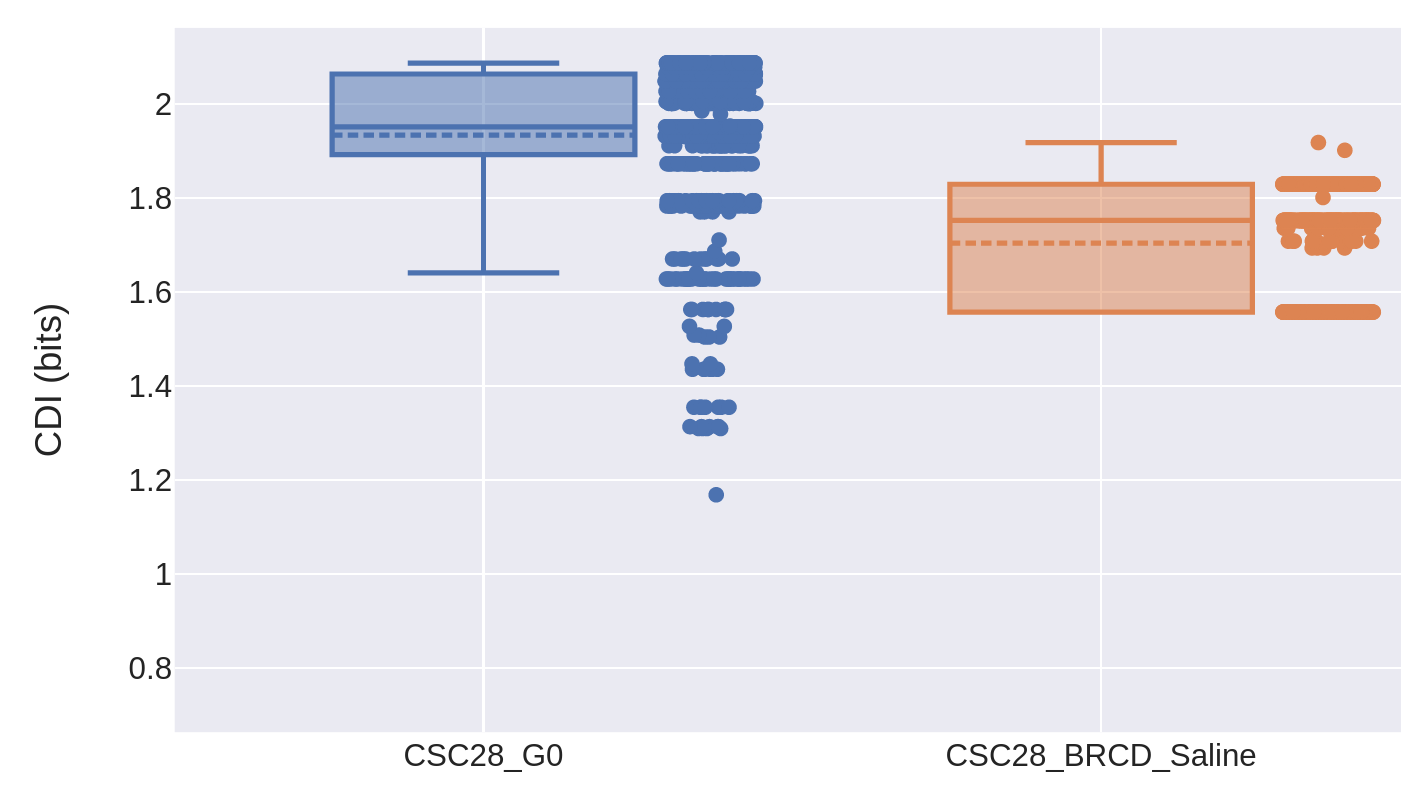
This paper, led by Dr. Catherine O'Brien's lab, showed that colorectal cancer cells can enter a reversible drug-tolerant persister state to evade death from chemotherapy and other targeted agents. I contributed to this project by using Pairtree to build clone trees for two cancers using multiple samples from each, then developing two information-theoretic measures to quantify the degree of genomic heterogeneity present in each cancer sample. This showed that multiple genomically distinct cell subpopulations were maintained in every tissue sample, including those taken before treatment, those that developed treatment resistance, and those that were seeded from the treatment-resistant samples. Though genomic heterogeneity was reduced in the treatment-resistant samples, it rebounded in the regrowth tumours seeded from them, lending support to the idea that drug tolerance arose through a non-genetic mechanism.
A practical guide to cancer subclonal reconstruction from DNA sequencing
January 2021Nature Methods
Tarabichi M, Salcedo A, Deshwar AG, Ni Leathlobhair M, Wintersinger J, Wedge DC, Van Loo P, Morris QD, Boutros PC.
This work provides an introduction to building subclonal reconstructions of cancer evolutionary history, reflecting the intricacies we learned through our participation in the Pan-Cancer Analysis of Whole Genomes project. I contributed material dealing with the analysis of copy-number aberrations, and how this affects downstream analyses.
Relapse-Fated Latent Diagnosis Subclones in Acute B Lineage Leukemia Are Drug Tolerant and Possess Distinct Metabolic Programs.
April 2020Cancer Discovery
Dobson SM, García-Prat L, Vanner RJ, Wintersinger J, Waanders E, Gu Z, McLeod J, Gan OI, Grandal I, Payne-Turner D, Edmonson MN, Ma X, Fan Y, Voisin V, Chan-Seng-Yue M, Xie SZ, Hosseini M, Abelson S, Gupta P, Rusch M, Shao Y, Olsen SR, Neale G, Chan SM, Bader G, Easton J, Guidos CJ, Danska JS, Zhang J, Minden MD, Morris Q, Mullighan CG, Dick JE.
Dr. John Dick's lab analyzed 14 B-progenitor acute lymphoblastic leukemias (B-ALLs), investigating the genomic and metabolic differences that allowed some subclones within a patient's cancer to tolerate treatment and seed disease relapse. To enable this work, his lab xenografted diagnosis and relapse samples from each patient into multiple mice and then subjected them to targeted sequencing, allowing us to analyze patient and mouse xenograft samples jointly as different depictions of a single cancer. This approach yielded up to 90 samples per cancer. My contribution was constructing clone trees for each patient, which revealed the genomic identities of the distinct subclonal populations composing a cancer, and allowed us to track their evolutionary trajectory between diagnosis and relapse. Additionally, by comparing the subclonal composition of mouse xenograft samples seeded from similar initial conditions, we gained insights into the stochasticity of cancer evolution. Existing methods for building clone trees failed when faced with such rich data, composed of up to 90 samples per cancer that revealed the existence of up to 26 subclonal populations. These failures forced us to consider why current approaches failed and how we could improve on them, leading to the creation of Pairtree.
The evolutionary history of 2,658 cancers
February 2020Nature
Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, Mitchell TJ, Rubanova Y, Anur P, Yu K, Tarabichi M, Deshwar A, Wintersinger J, Kleinheinz K, Vázquez-García I, Haase K, Jerman L, Sengupta S, Macintyre G, Malikic S, Donmez N, Livitz DG, Cmero M, Demeulemeester J, Schumacher S, Fan Y, Yao X, Lee J, Schlesner M, Boutros PC, Bowtell DD, Zhu H, Getz G, Imielinski M, Beroukhim R, Sahinalp SC, Ji Y, Peifer M, Markowetz F, Mustonen V, Yuan K, Wang W, Morris QD, Spellman PT, Wedge DC, Loo PV.
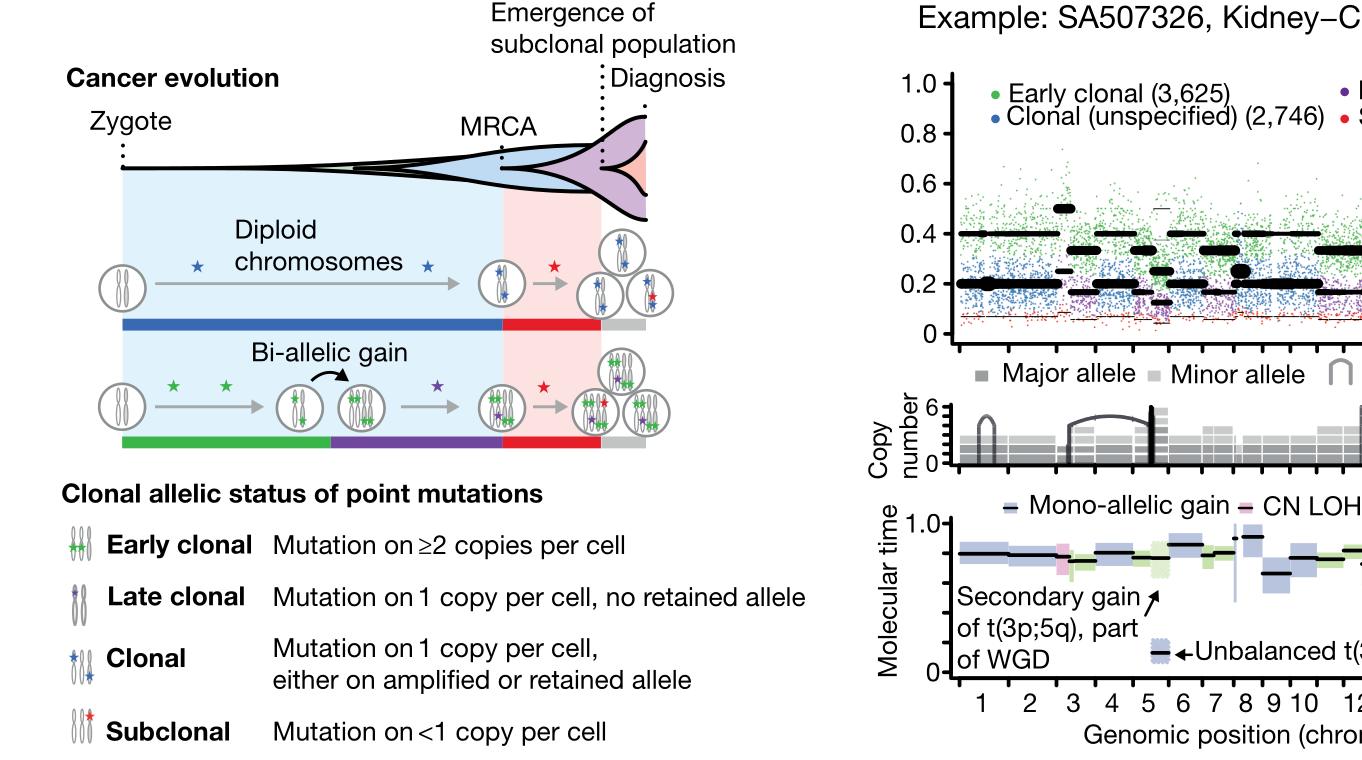
This was the second major paper to emerge from our Evolution and Heterogeneity working group within the Pan-Cancer Analysis of Whole Genomes. Building on the characterizations of genomic evolution established by the other paper (described above), this work examined the timing of major events in cancer evolution, finding that oncogenesis is characterized by mutations in a small number of driver genes, while later tumour evolution is driven by mutations in a much broader gene set. Additionally, the study found that foundational genomic alterations critical to some cancers, such as whole-genome duplications, can precede disease onset by years or decades. I contributed to this work both through creating the consensus copy-number profiles described above, and by using my lab's methods for reconstructing cancer evolutionary history to contribute to the group's consensus evolutionary profiles for each cancer.
A community effort to create standards for evaluating tumor subclonal reconstruction
January 2020Nature Biotechnology
Salcedo A, Tarabichi M, Espiritu SMG, Deshwar AG, David M, Wilson NM, Dentro S, Wintersinger JA, Liu LY, Ko M, Sivanandan S, Zhang H, Zhu K, Yang T-HO, Chilton JM, Buchanan A, Lalansingh CM, P’ng C, Anghel CV, Umar I, Lo B, Zou W, Simpson JT, Stuart JM, Anastassiou D, Guan Y, Ewing AD, Ellrott K, Wedge DC, Morris Q, Loo PV, Boutros PC.
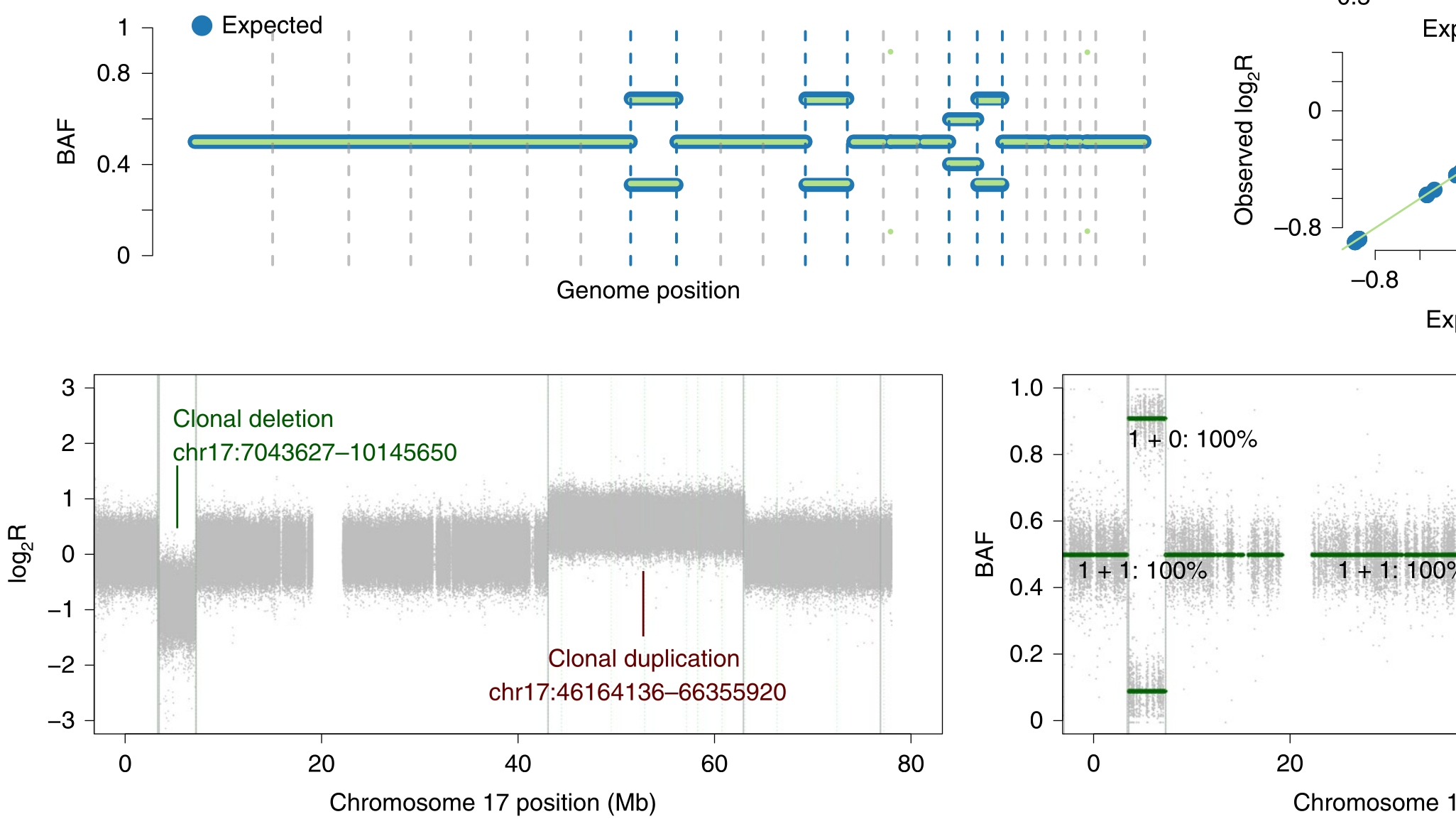
This paper developed several novel means of comparing cancer subclonal reconstruction methods, then benchmarked leading methods to see how well they could recover a known truth from simulated data. I contributed by assisting with the benchmarking.
Old papers
Projects from my undergraduate research
One species, two genomes: A critical assessment of inter isolate variation and identification of assembly incongruence in Haemonchus contortus
August 2018bioRxiv
Wintersinger J, Mariene GM, Wasmuth J.
For my bachelor's thesis, I characterized two published reference genomes for the parasitic nematode Haemonchus contortus. While some differences resulted from legitimate biological differences between the two divergent isolates that were sequenced to produce these genomes, others stemmed from technical deficiencies in assembly or annotation of one or both genomes. I developed several approaches for disentangling what discrepancies arose from biological variation, and what came from technical error. Notably, only 45% of genes in one genome were orthologous to genes in the other, with one genome exhibiting almost as much orthology to C. elegans as its counterpart H. contortus strain, suggesting considerable error in one or both genomes. While the nematode research community has since published a new H. contortus reference genome that overcame many of the issues enumerated in this work, my project serves as an illustration of the technical errors that can occur in building reference genomes, and how they can grossly distort a reference genome's representation of biological reality.
Kablammo: an interactive, web-based BLAST results visualizer
April 2015Bioinformatics
Wintersinger JA, Wasmuth JD.
As part of my bachelor's thesis, I developed Kablammo, a web-based tool for visualizing BLAST results. The method has been used by a number of other studies to produce figures for publication. As an extension of this work, I developed a dynamic programming algorithm for computing the optimal set of BLAST hits.
Other papers
Projects to which I made minor contributions
| Title | Date | Journal | Authors |
|---|---|---|---|
| Reconstructing evolutionary trajectories of mutation signature activities in cancer using TrackSig | February 2020 | Nature Communications | Rubanova Y, Shi R, Harrigan CF, Li R, Wintersinger J, Sahin N, Deshwar A, Morris Q |
| The Evolutionary Landscape of Localized Prostate Cancers Drives Clinical Aggression | May 2018 | Cell | Espiritu SMG, Liu LY, Rubanova Y, Bhandari V, Holgersen EM, Szyca LM, Fox NS, Chua MLK, Yamaguchi TN, Heisler LE, Livingstone J, Wintersinger J, Yousif F, Lalonde E, Rouette A, Salcedo A, Houlahan KE, Li CH, Huang V, Fraser M, van der Kwast T, Morris QD, Bristow RG, Boutros PC |
| An undergraduate perspective on LINDSAY Composer: Bridging the gap between software engineering and physiology | July 2013 | Journal of Undergraduate Research in Alberta | Yuen DWK, Karaman T, Wintersinger J |